The century-old Haber-Bosch process is responsible for 90% of the annual industrial production of ammonia, a vital component of fertilizers for agriculture. However, this process is extremely energy-intensive and inefficient, consuming 2% of global energy and releasing 400 Mt of CO2 per year to satisfy current demands. Thus, a need of the hour for sustainable food production is to find alternative, greener methods of reducing nitrogen. One such alternative process is electrocatalysis, where a specially-designed material catalyzes the decomposition of nitrogen on its surface, often with far lower energy requirements and a much smaller carbon footprint.
Academic pursuits towards finding novel electrocatalyst materials have so far mostly focused on optimizing the activity metric (i.e., how reactive the material is towards the target reaction). However, to find a commercially-viable electrocatalyst material, several other, often conflicting, factors need to be considered. For example, the candidate material should not be prohibitively expensive to make, and must remain stable during usage in relevant operating conditions. Thus, efforts to design new electrocatalysts must take into account these competing objectives and constraints to yield feasible results.
Extending the state-of-the-art
To tackle this challenge, we recently prototyped a framework for finding new electrocatalysts while simultaneously optimizing conflicting objectives such as activity, stability, and cost. We integrated this framework into an autonomous sequential learning (SL) workflow that helped explore a vast design space of potential electrocatalyst candidates and quickly identify the most-promising ones to assess using high-fidelity atomistic simulations.
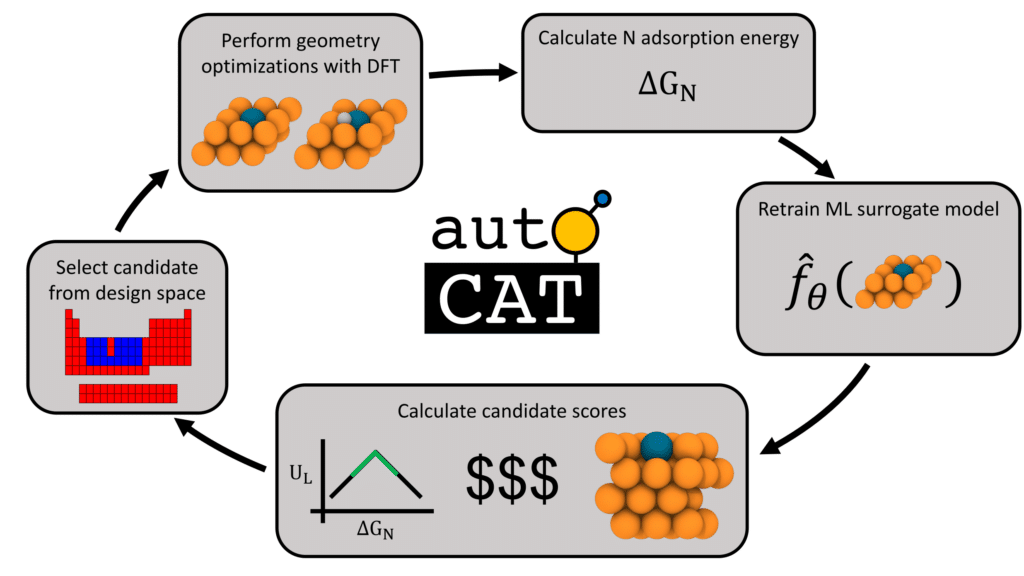
New electrocatalysts identified!
The above approach was used to search for electrocatalysts in a new class of materials, called single-atom alloys (SAAs). We successfully identified several hitherto-unknown potentially high-performing materials and validated them using quantum mechanical calculations, all within a handful of SL iterations. This work was done in collaboration with partners at Carnegie Mellon University, as part of a US Department of Energy ARPA-E initiative called DIFFERENTIATE.
Read More
If you are interested in finding out more about these findings, a preprint of our manuscript can be found in the ChemRxiv repository at this URL: